Genomics | Transcriptomics | Genotyping
Analyseverfahren | Analysis Workflows
Einige der verfügbaren Analyseverfahren sind im folgenden erläuter. Wenn Sie weitere Informationen benötigen, kontaktieren sie bitte Dr. Alfons Weig.
Some of the available procedures are explained below. If you seek more information, please contact Dr. Alfons Weig.
- Amplicon SequencingEinklappen
-
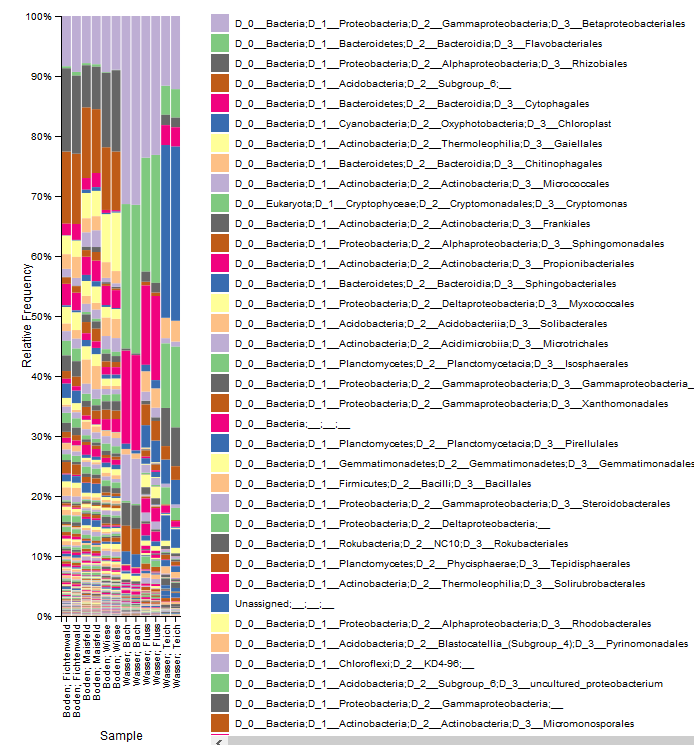
© Alfons Weig
The Amplicon-Seq method is used to investigate the microbial community composition with next-generation sequencing technologies. The most often used phylogenetic loci are:
- 16S rDNA (bacteria)
- 18S rDNA (eucaryotes)
- ITS (mainly fungal communities)
The analysis of other phylogenetic loci is also possible, but should be discussed in detail for each experiment.
- 16S rDNA (bacteria)
- Transcriptom SequencingEinklappen
-
RNA-Seq methods enable us to determine the transcript amounts of genes in an organism, or in tissues, organs, and very limiting material like single cells. A comparison of transcriptome profiles between samples enables us to identify differentially-expressed genes which could play an important role in biological processes.
In addition, RNA-Seq allows the identification of splice variants, that occur under certain conditions.
RNA-Seq experiments can be conducted at different sequencing depths, depending on the genomic complexity of the underlying genomen (e.g. size, ploidy levely) and an the library type.
- Genome SequencingEinklappen
-
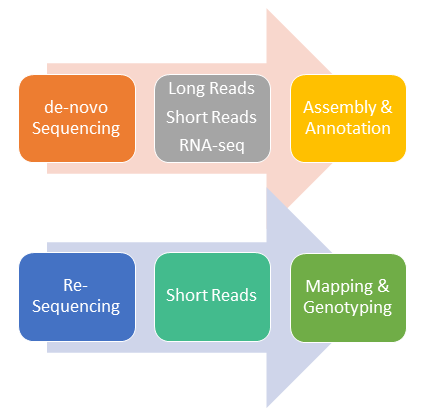
© Alfons Weig
De-novo genome sequencing of non-model organisms is often perforemd by a combination of different sequencing technologies: Long-read sequencing data (e.g. Nanopore, PacBio) can be combined with short-read data (e.g. Illumina) to assemble large scaffolds of genomic sequences at very high sequence quality. RNA-Seq data will then be combined with the draft genome assembly to support gene prediction and functional annotation of putative genes.
On the other hand, re-sequencing of genomes from organisms with an existing reference genome allow the identification of genomic variations in strains and/or mutants of this organims.
The selection of the most appropriate sequencing technology is dependent on the genomic complexity (genome size) of the respective organism.
- GenotypingEinklappen
-
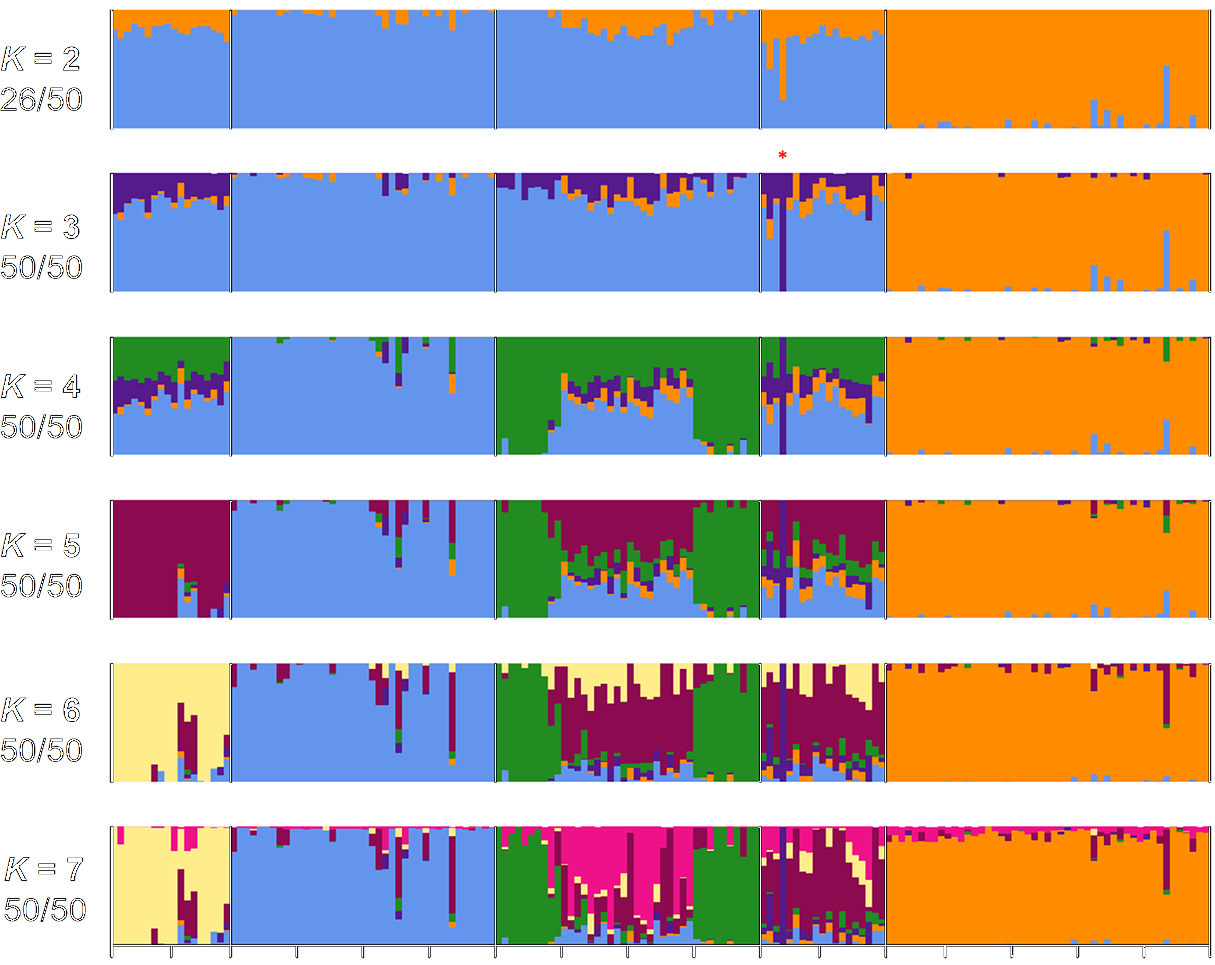
© DOI: 10.1371/journal.pone.0177742
The identification of genotypic variations within a species or between closely-related species is an important aspect for functional genomic analyses. Genetic variations form the molecular basis for adaptation to environmental changes, yield, stress tolerance and other processes. Genotyping experiments can be conducted at very different resolution, ranging from the analysis of several 100s to 10,000s or millions of genetic markers.
Genotyping-by-Sequencing (e.g. RAD-Seq, nextRAD-Seq) is based on high-throughput sequencing of representative regions of genomes and delivers several 10,000s of genetic markers for population studies.
ISSR analyses are more simple methods for genotyping at low resolution.
- Fingerprinting of Communities Einklappen
-
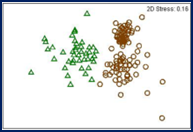
© DOI: 10.1007/s11557-012-0883-1
The ARISA method represents a quick and low-cost alternative to Amplicon-Seq. It allows the anonymous identification of community dynamics on a qualitative level. ARISA is mainly applied for bacterial and fungal communities.
- Population GeneticsEinklappen
-
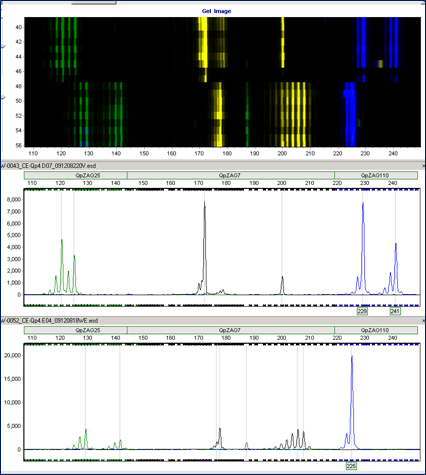
© Alfons Weig
Microatellite analyses enable us to investigate the composition and dynamics of populations by fingerprinting methods.
Microsatellite marker - either from published studies, or from de-novo marker development - must be checked for their usability (e.g. polymorphism) in the respective experiments.